What Is High-Throughput RNA Validation?
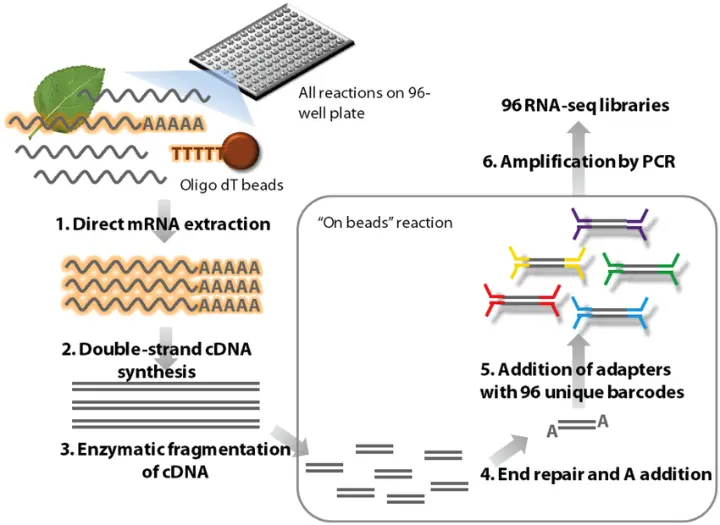
High-throughput RNA validation is a critical step in the gene expression workflow, used to confirm results obtained from large-scale transcriptomic studies such as RNA sequencing (RNA-Seq). While RNA-Seq is powerful for discovery, its data often require validation through more targeted and quantitative methods. High-throughput validation ensures that key expression changes are real, reproducible, and biologically meaningful.
How High-Throughput RNA Structure Probing Works ?
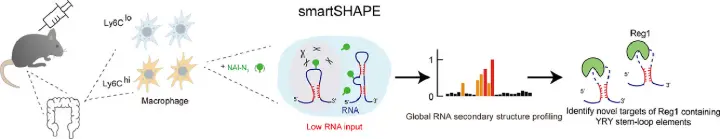
High-throughput RNA structure probing allows researchers to explore RNA folding across the entire transcriptome. This method uses chemical or enzymatic reagents that selectively modify or cut single-stranded regions of RNA, leaving molecular marks. During reverse transcription, these marks cause either stops or mutations in the cDNA, which are then detected using next-generation sequencing.
By analyzing the frequency and location of these modifications, scientists can map RNA flexibility and predict structural patterns. Unlike traditional techniques, this approach can be applied to thousands of RNAs at once even directly in living cells offering insight into how RNA structure influences function under different biological conditions.
A common conceptual core for determining RNA structures with high-throughput sequencing
The Role of RT-qPCR in Validating High-Throughput RNA Data
Reverse transcription quantitative PCR (RT-qPCR) is widely regarded as the gold standard for validating high-throughput RNA expression data obtained from methods like RNA-Seq and microarrays. These large-scale techniques are excellent for generating comprehensive transcriptional profiles, but they often require independent validation to confirm differential expression and to rule out technical artifacts. RT-qPCR offers the necessary sensitivity, specificity, and dynamic range to reliably verify expression changes in selected genes of interest.
From Sequencing to Structural Maps
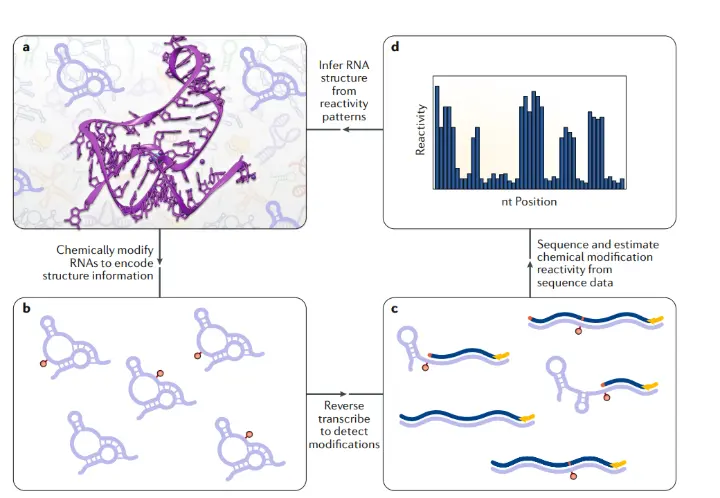
Once high-throughput RNA structure probing experiments are completed, the vast amount of sequencing data generated must be analyzed using specialized bioinformatics pipelines. These computational tools align the reads to reference transcripts and identify positions where chemical modifications or enzymatic cuts occurred signals that reflect RNA flexibility at the nucleotide level. The result is a reactivity profile, a quantitative map showing how accessible or constrained each nucleotide is within the RNA molecule.
These reactivity profiles are then integrated into secondary structure prediction algorithms, which model base-pair interactions and folding patterns. Unlike traditional sequence-only approaches, this structure-informed modeling provides a more accurate representation of RNA conformations in their native environment. With this approach, researchers can analyze thousands of RNAs simultaneously, revealing how structural elements affect key biological functions such as mRNA stability, translation regulation, splicing, and RNA-protein interactions.
By capturing the dynamic nature of RNA folding, these tools uncover regulatory features that would otherwise be hidden in sequence data alone, offering new insights into both normal cellular processes and disease mechanisms.